omics4plant
Dataget: get anndata/seurat from matrix
Dataget-SoupX
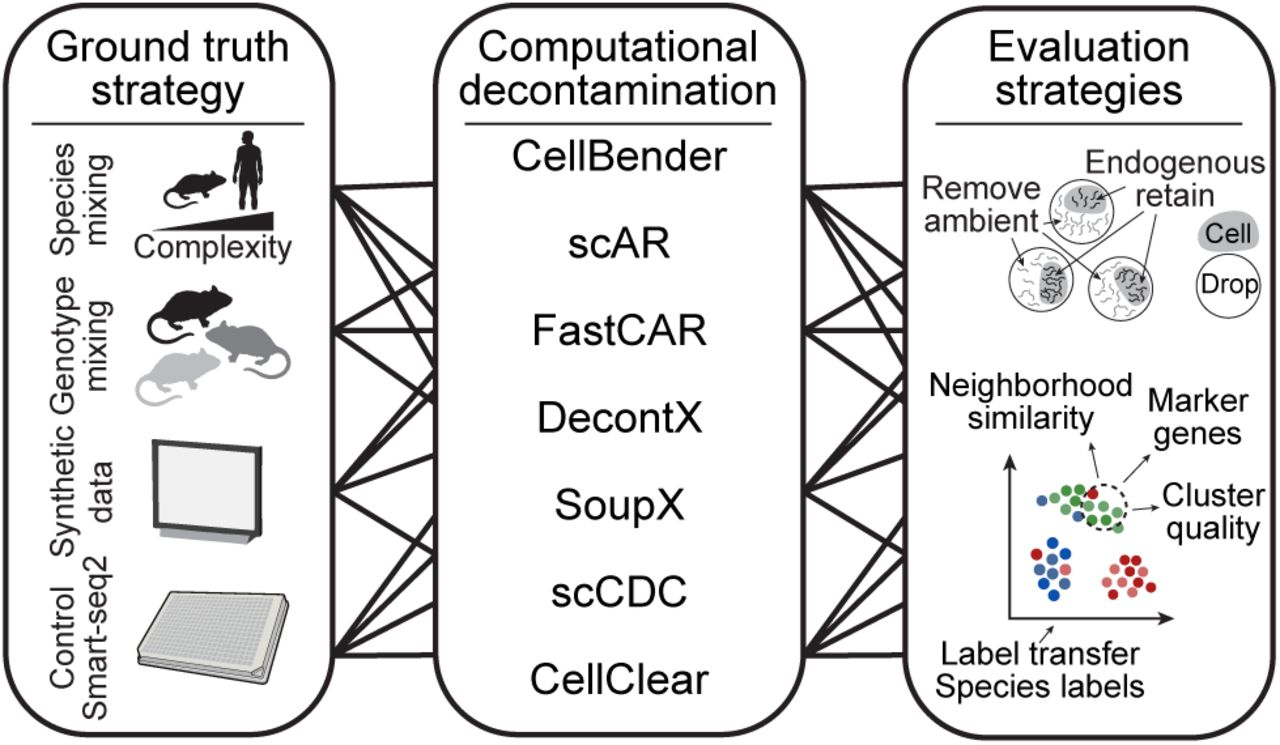
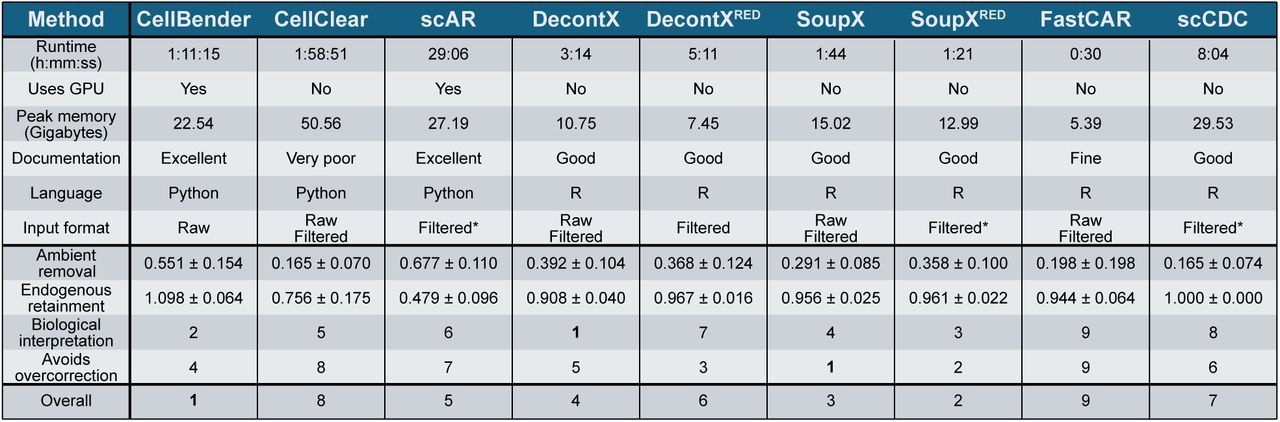
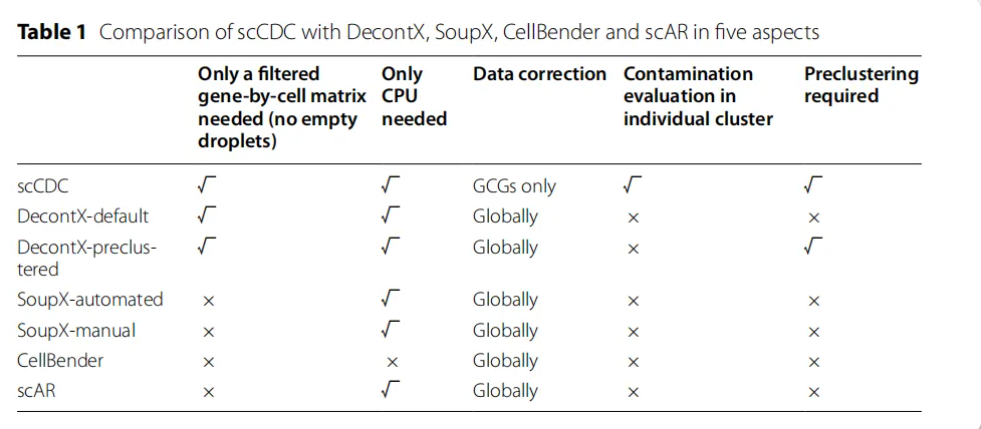
搭建一个多方法RNA去污染的R包,包括SoupX,decontx,scCDC和FastCAR。支持两种模式输入,一种模式包含raw和filter矩阵,一种是只有filter矩阵。另外附带一些数据统计和可视化方案,最终输出矫正后的矩阵和可视化。输入文件:1)raw+filter;2)filter。是否聚类:1)no 2)need。评估去污:1)ambient RNA的去除;2)生物学性状的保留:cluster、marker、annotation。横向对比:corrected与uncorrected的对比。
- Variables
rawMatrix(Required, default to None) 原始表达矩阵filterMatrix(Required, default to None) 过滤后表达矩阵prefix(Required, default to None) 文件输出后缀min_genes(Required, default to 100) 过滤少于min_genes基因表达的细胞min_cells(Required, default to 3) 过滤少于在min_cells细胞表达的基因tfidfMin(Required, default to 1) Minimum value of tfidf to accept for a marker gene.roundToInt(Required, default to true) SoupX输出的矩阵为整数矩阵mem_Decontamination(Required, default to 32) 内存使用G
- Softwares
- SoupX
decontxscCDCFastCAR
- Output
- env
source /opt/software/miniconda3/bin/activate conda config --add channels conda-forge conda create -n R4.3 r-base=4.3 -y conda activate R4.3 conda install conda-forge::r-devtools -y conda install conda-forge::r-seurat -y conda install conda-forge::r-ddpcr -y conda install conda-forge::r-proc -y Rscript -e 'devtools::install_github("ZJU-UoE-CCW-LAB/scCDC")' # scCDC conda install bioconda::bioconductor-decontx -y # DecontX conda install conda-forge::r-soupx -y # SoupX Rscript -e 'devtools::install_git("https://git.web.rug.nl/P278949/FastCAR")' # FastCAR # devtools::install_github("immunogenomics/presto") conda install bioconda::bioconductor-dropletutils -y conda install conda-forge::r-optparse -y conda install conda-forge::r-reticulate -y conda install conda-forge::r-irkernel -y conda install bioconda::bioconductor-scdblfinder -ylapply(c("Seurat","DropletUtils","SoupX", "optparse", "decontX", "FastCAR", "scCDC", "qlcMatrix", "Matrix", "scater"), library, character.only = T)
Dataget-FilterDoublet
- Brief
- Variables
- Softwares
- Output
- env
# devtools::install_github("cellgeni/schard") conda create -n py python=3.12 -y conda activate py conda install conda-forge::scanpy -y conda install bioconda::scrublet -y conda install conda-forge::leidenalg -y